데이터 살펴보기 - GEO series 데이터 자세히 확인하는 법
안녕하세요 한헌종입니다.
이전 포스트에서는 GEO 에서 원하는 데이터를 어떻게 찾는지 설명드렸습니다.
오늘은 원하는 데이터를 찾은 뒤 이 데이터가 사용 가능한지 확인하는 방법을 설명드리겠습니다.
물론 GEO 는 open source 이므로 업로드 되어있는 데이터는 모두 사용 가능합니다.
그럼 데이터가 사용 가능한지를 왜 체크해야 할까요?
이는 여러분들이 원하는 형태와 종류의 데이터가 있을수도, 없을수도 있기 때문입니다.
1. 데이터 종류 확인
여러분이 먼저 확인해야 할 정보는 바로 데이터가 어떤 종류인지 확인하는 겁니다.
이 시리즈 데이터가 Microarray 데이터인지 bulk RNA-seq data 인지 single-cell RNA-seq 데이터인지 알아야 쓸지말지 결정할 수 있겠죠?
보통은 title 에도 나와있을 수 있지만, 더 자세히 확인하는 방법은
- Experiment type 항목 확인
- Platform 항목 확인
이 두가지가 될것 같습니다.
예시로 GSE101108 샘플을 한번 살펴볼까요?
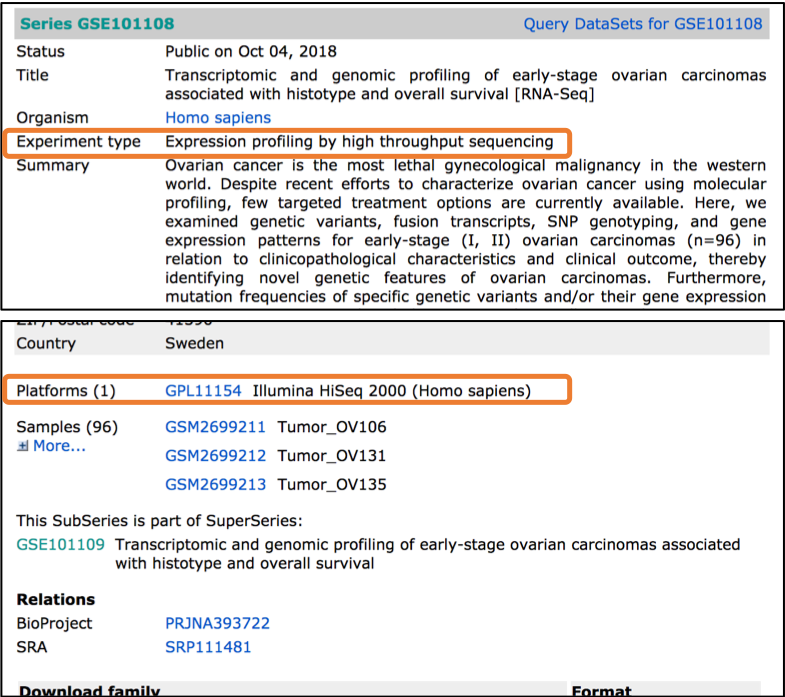 |
이 시리즈에서는 Experiment type 이 다음과 같습니다.
- Expression profiling by high throughput sequencing
또한 platform 정보는 다음과 같습니다.
- GPL11154 Illumina HiSeq 2000 (Homo sapiens)
즉, 이 시리즈에는 시퀀싱 플랫폼을 사용한 RNA-seq 데이터가 있는 것입니다.
더 자세한 내용은 나머지 experimental design 이나 summary, 그리고 나중에 말씀드릴 series matrix 를 살펴보시면 됩니다.
여러분이 원하는 데이터 종류가 맞는지 꼭 확인하세요!
2. 제공하는 데이터 형식 확인
자, 원하는 데이터 종류가 맞다면 그 다음으로 해야할 것이 바로 원하는 형식의 데이터를 제공해주는가 입니다.
만약 여러분께서 각 sample 마다 각 유전자의 발현량이 적혀있는 expression matrix 를 원하신다면, 이 시리즈 데이터가 그것을 제공하는지 꼭 확인해봐야 합니다.
그 이유는, 보통 Microarray 를 사용한 expression data 는 발현량 intensity data 를 제공하지만, RNA-seq 데이터는 제공하지 않는 경우도 많거든요.
또한 여러분께서 RNA-seq raw data 인 fastq 파일을 원하신다면, 이 또한 제공되는지 확인해봐야 합니다.
자, 그럼 데이터를 제공하는지는 어떻게 알 수 있을까요?
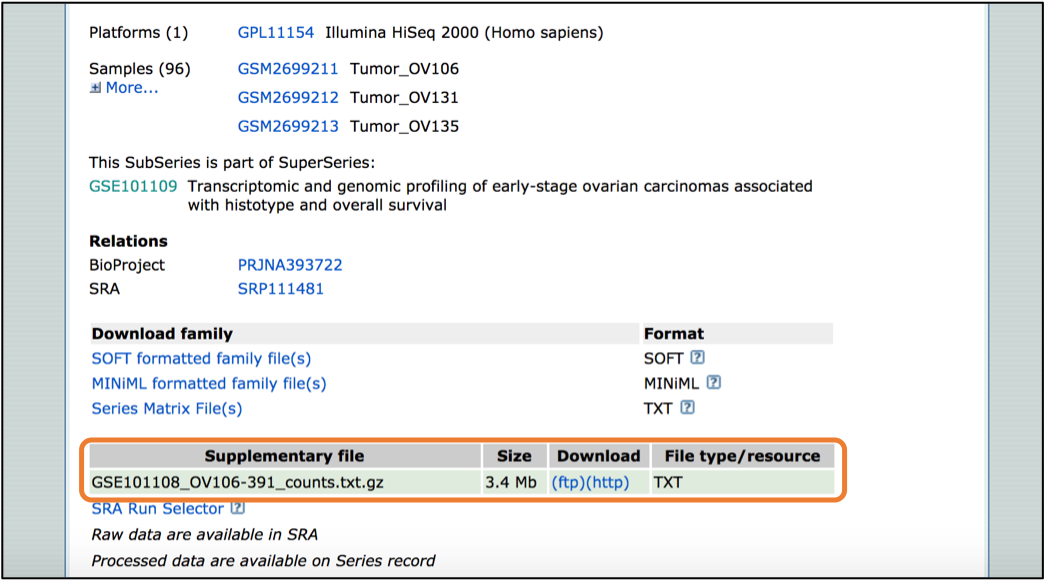
바로 시리즈 데이터 아래쪽에 나온 Supplementary file 부분을 확인하시면 됩니다.
각 파일마다 Download 컬럼이 있는데, 보시면 http, ftp, custom 등이 적혀있을 겁니다.
GSE101108 의 경우를 보시면 http, ftp 두개가 있군요.
 |
http 혹은 ftp 를 누르시면 그 파일이 바로 다운로드 받아질 겁니다.
custom 이 있는 경우 이를 눌러보면, 만약 그 파일이 여러 파일이 모여있는 거라면 어떤 파일들이 있는지 확인 가능합니다.
또한 그 파일들 중 일부만 선택해서 다운로드 받을 수 있습니다.
그럼 사용 가능한 파일이 있는지 확인해볼까요?
다운로드 가능한 파일이 .txt.gz 형식이면서 크기는 3.4 Mb 이고, 제목에는 _count 라고 되어있군요.
아마도 이 데이터의 저자들이 이미 분석해놓은 expression matrix 인것 같습니다.
다만 FPKM, TPM 이 아닌 count 데이터이므로 normalization 과정이 필요하겠군요.
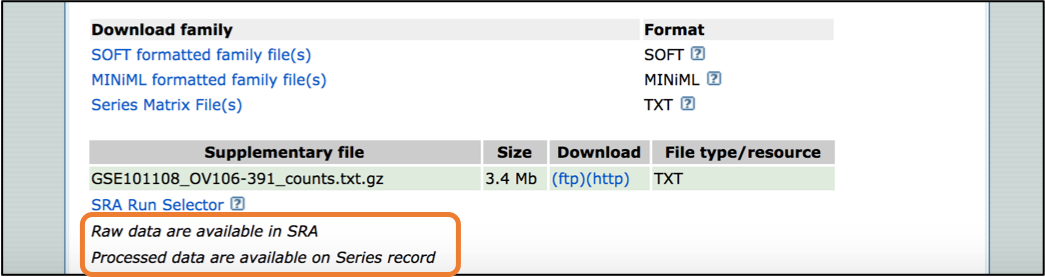 |
맨 아래의 글귀를 확인해보셔도 제공되는 데이터를 확인하실 수 있습니다:
Raw data are available in SRA: SRA 라는 링크를 통해 fastq 와 같은 raw data 를 받을 수 있다는 뜻입니다.
Processed data are available on Series record: 어느정도 분석이 완료된 Expression matrix (여기서는 count 로 된 데이터) 가 제공된다는 뜻입니다.
어떤 시리즈에서는 이렇게 expression matrix, 즉 processed data가 제공되지 않는 경우도 있습니다.
raw data 만 제공되는 경우인데요, 이런 경우 직접 fastq 파일을 받아서 분석하시는 방법밖에는 없습니다.
데이터 분석에 익숙하지 않은 분이라면 이런 데이터는 사용하기 힘들 수 있을 거에요.
그렇기 때문에 이 방법으로 제공되는 데이터의 종류를 확인하시는게 무척 중요합니다!
3. 데이터를 더욱 자세히 확인하는 방법: Series matrix
데이터가 어디에서 온 샘플이고, 어떻게 만들어졌으며, 어떤 방식으로 분석되었는지 자세히 알고 싶으신가요?
이런 자세한 정보는 바로 series matrix 라는 데이터에 잘 적혀 있습니다.
사실 이 파일은 저자가 GEO 에 데이터를 업로드하기 위해 꼭 작성해야 하는 파일 중 하나입니다.
얼마나 자세히 적을지는 저자에게 달려있지만, 왠만한 정보는 다 적혀있다고 보시면 됩니다.
또한, Microarray 데이터의 경우에는 이 파일에 Expression matrix 까지 적혀있습니다!
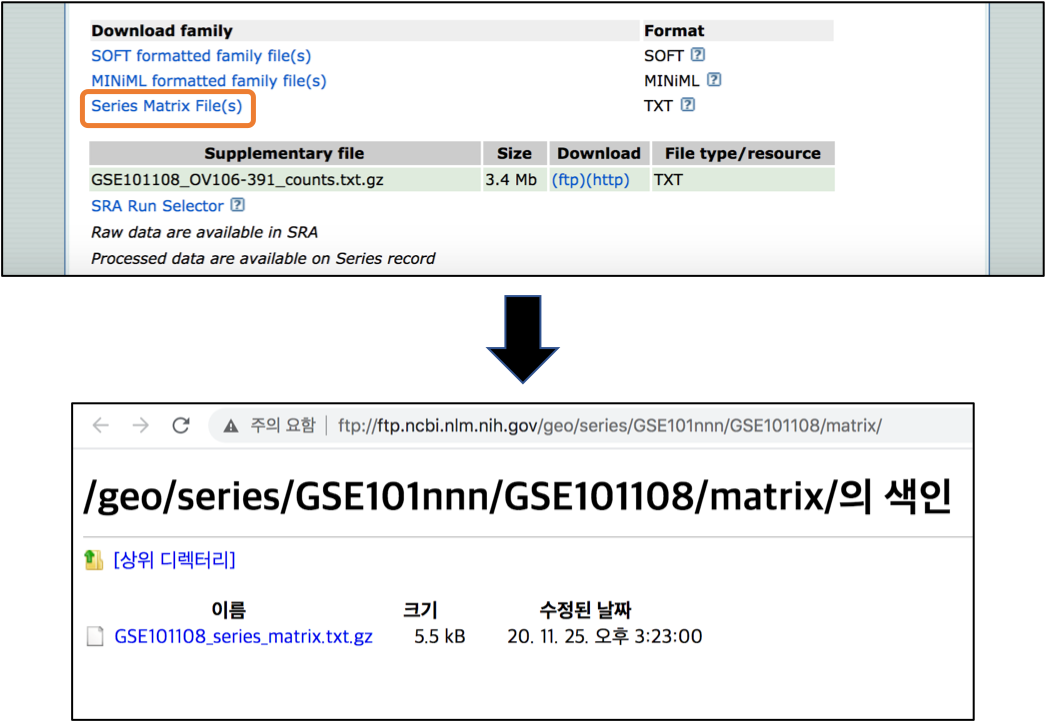
자, 그럼 series matrix 는 어디서 받을 수 있을까요?
바로 시리즈 데이터 아래쪽 Download family 중에 있습니다.
이중 Series Matrix File(s) 를 선택하시면 이를 받을 수 있는 ftp site 로 넘어갑니다.
 |
다운로드 받으신 데이터는 txt 파일이긴 한데, 실제로는 tsv 파일 (탭으로 나누어진 파일) 입니다.
좀 더 편하게 보시려면 Excel 을 통해 열어보시는걸 추천드립니다.
엑셀로 열어보시면, 다음과 같이 데이터의 자세한 정보가 표 형태로 적혀있을 겁니다.
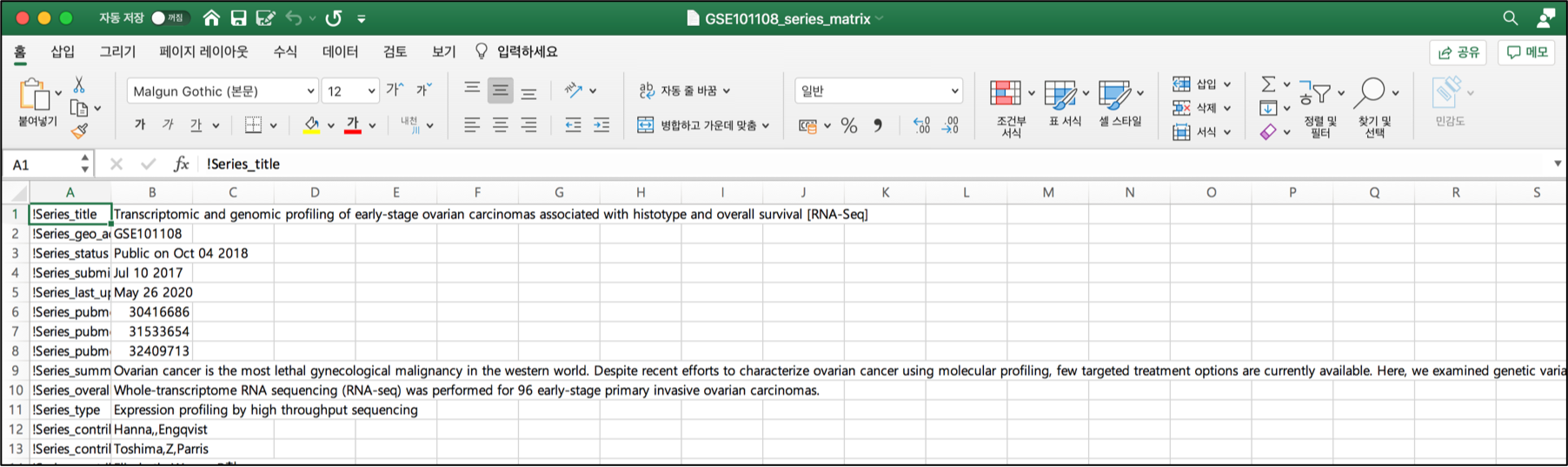 |
Series matrix 는 크게 두 부분으로 나눌 수 있습니다.
바로 시리즈에 대한 정보와 샘플에 대한 정보 입니다.
위쪽에는 시리즈에 대한 정보가 나타납니다.
아까 해당 시리즈 창에서 확인하셨던 데이터가 주로 여기에 그대로 나타날 겁니다.
GEO accession ID, Submission date, Summary 등이 바로 그 정보입니다.
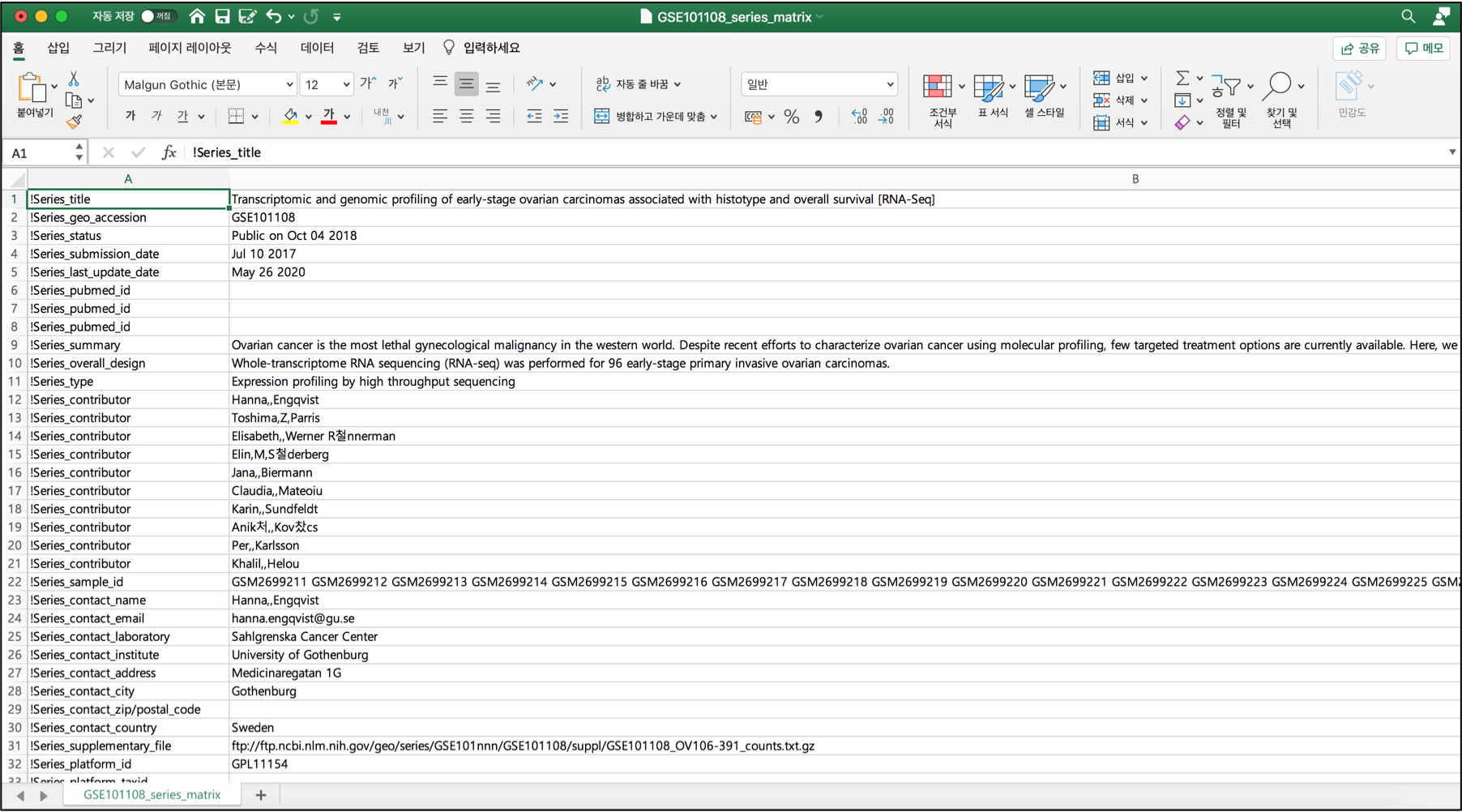 |
그리고 아래쪽에는 각 샘플에 대한 정보가 적혀있습니다.
여기에는 샘플이 어디서 왔는지, 어떤 culture 조건으로 키웠는지, 사용한 media 는 무엇인지, treatment 는 언제 어떻게 했는지 등이 적혀 있습니다.
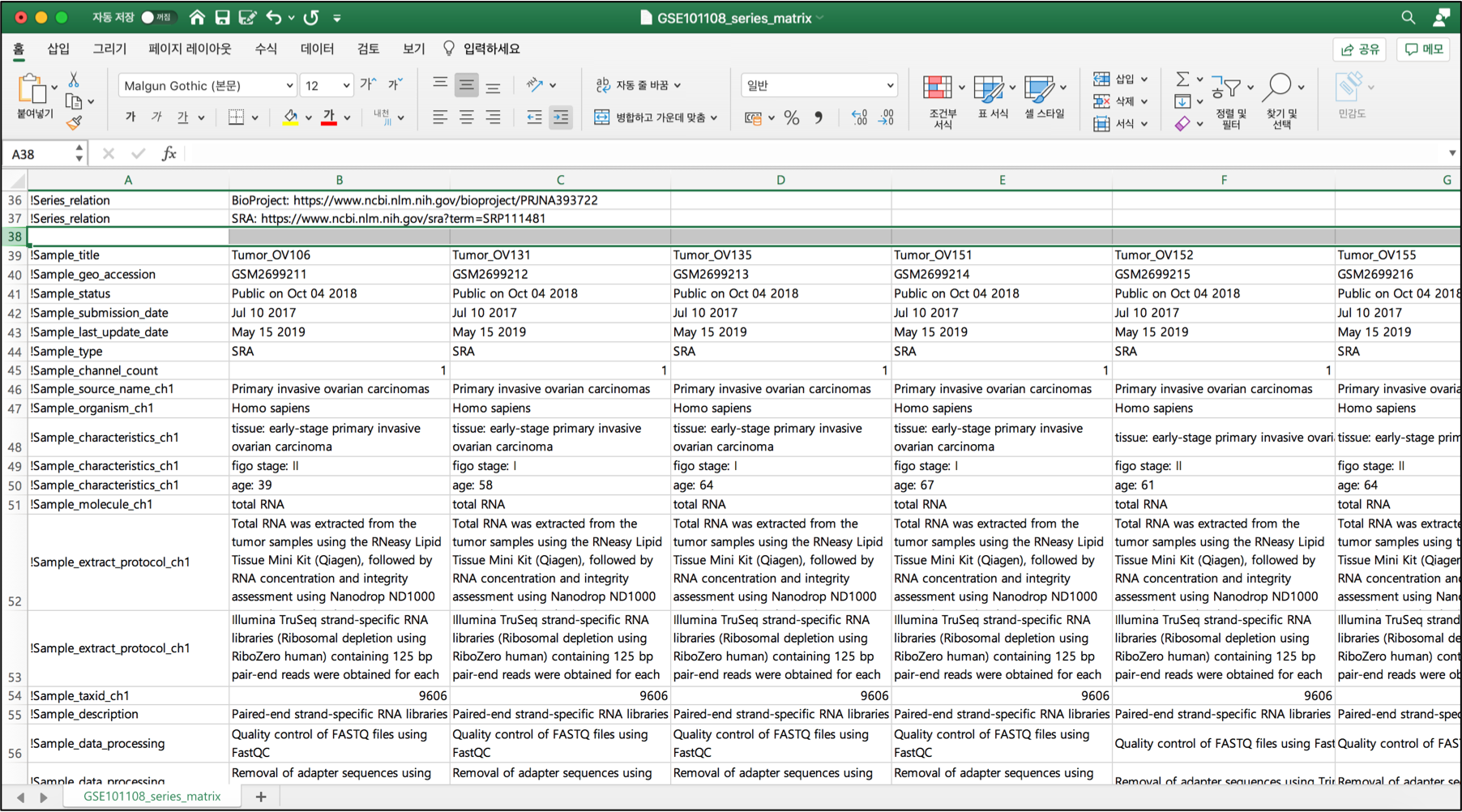 |
예를 들어 첫 번째 샘플을 볼까요? (GSM2699211)
다음 정보들이 적혀있습니다.
early-stage primary invasive ovarian carcinoma 조직에서 가져왔고, 환자 나이는 39세 인 것을 알 수 있습니다.
| !Sample_title | Tumor_OV106 |
| !Sample_geo_accession | GSM2699211 |
| !Sample_status | Public on Oct 04 2018 |
| !Sample_submission_date | Jul 10 2017 |
| !Sample_last_update_date | May 15 2019 |
| !Sample_type | SRA |
| !Sample_channel_count | 1 |
| !Sample_source_name_ch1 | Primary invasive ovarian carcinomas |
| !Sample_organism_ch1 | Homo sapiens |
| !Sample_characteristics_ch1 | tissue: early-stage primary invasive ovarian carcinoma |
| !Sample_characteristics_ch1 | figo stage: II |
| !Sample_characteristics_ch1 | age: 39 |
| !Sample_molecule_ch1 | total RNA |
또한 이 샘플의 데이터를 어떻게 분석했는지에 대한 정보도 매우 자세하게 나오고 있습니다.
첫 번째 샘플은 다음과 같이 분석되었군요.
STAR 툴을 사용해 read alignment 를 진행했고, HTSeq 툴을 써서 raw read count를 계산했다고 합니다.
그리고 Reference genome 은 hg19 를 사용했네요.
| !Sample_description | Paired-end strand-specific RNA libraries |
| !Sample_data_processing | Quality control of FASTQ files using FastQC |
| !Sample_data_processing | Removal of adapter sequences using TrimGalore wrapper script |
| !Sample_data_processing | FASTQ files were mapped to the human reference genome hg19 using the STAR aligner and the resulting BAM-file was name-sorted |
| !Sample_data_processing | Raw read counts were calculated using htseq-count in htseq |
| !Sample_data_processing | Genome_build: hg19 |
| !Sample_data_processing | Supplementary_files_format_and_content: The processed data file “OV106-391_counts.txt” is given in text format |
그리고, 마지막으로 샘플을 분석한 기관 및 연락처가 나와있습니다.
스웨덴의 Sahlgrenska Cancer Center, University of Gothenburg 에서 만들어진 샘플이네요.
이 외에도 해당 샘플의 raw data 를 어디서 받을 수 있는지도 나와있군요.
| !Sample_contact_name | Hanna,,Engqvist |
| !Sample_contact_email | hanna.engqvist@gu.se |
| !Sample_contact_laboratory | Sahlgrenska Cancer Center |
| !Sample_contact_institute | University of Gothenburg |
| !Sample_contact_address | Medicinaregatan 1G |
| !Sample_contact_city | Gothenburg |
| !Sample_contact_zip/postal_code | 41390 |
| !Sample_contact_country | Sweden |
| !Sample_data_row_count | 0 |
이렇게 각 샘플의 정보를 자세히 살펴보면, 이 데이터가 지금 내가 원하는 조직의 샘플인지, 원하는 실험 조건으로 만들어진 샘플인지 확인할 수 있습니다.
또한 이 series matrix 의 정보를 통해 샘플들을 특정 조건으로 대조군, 비교군으로 나눌 수 있습니다.
Series matrix 는 길고 복잡하고 보기 어렵게 되어있지만, 유용한 정보가 많으니 시간이 된다면 확인해보시는게 좋아요.
네, 오늘은 GEO series 데이터를 어떻게 자세히 살펴볼 수 있는지 설명해봤습니다.
이 글을 참고하셔서 GEO 데이터를 사용하실 때 좀 더 쉽고 효율적으로 원하는 데이터를 찾고 사용하시길 바랄게요.
그럼 다음 시간에 만나요!