RNA-seq raw data 부터 분석하는 방법 - 서버 없이!
안녕하세요, 한헌종입니다.
오늘은 RNA-seq 데이터를 raw data 부터 분석하는 법을 설명해보고자 합니다.
RNA-seq 데이터는 GEO 같은 곳에 넘쳐나는데 분석하기 힘드신 분들 계실겁니다.
또한 실험을 통해 RNA-seq 데이터는 받았는데, raw 데이터만 제공받아서 ‘이걸 어떻게 해야하지’ 하는 분들 많으실 겁니다.
이런 경우는 리눅스를 다를 줄 모르시거나, 적절한 사양의 서버가 없거나, 다양한 이유가 있을 거에요.
오늘은 간단하게, 리눅스 명령어 없이, 서버 없이 RNA-seq 데이터를 raw-data 부터 분석하는 법을 말씀드릴 겁니다!
조금 길더라도 천천히 따라해보시면 여러분도 쉽게 해보실 수 있습니다.
 |
|---|
| 원래는 이런 까만 화면을 통해 리눅스 명령어를 입력해가며 분석해야 하는 일이지만, 오늘은 필요 없어요 |
Galaxy 서버를 소개합니다.
자, 어떻게 서버 없이, 명령어 없이 시퀀싱 데이터 분석이 가능한 걸까요?
이는 Galaxy 라는 클라우드 사이트가 있기 때문에 가능합니다.
주소는 다음과 같습니다: https://usegalaxy.org/
이 Galaxy 라는 서버는, 사용자가 여러 시퀀싱 데이터 및 기타 데이터를 업로드해서 다양한 분석을 할 수 있는 곳입니다.
한번 접속해서 어떻게 생겼는지 확인해보세요!
 |
|---|
| galaxy 홈페이지 입니다. |
이제 이곳을 통해 여러분이 원하시는 분석을 할 수 있습니다.
사용하시기 전에 회원가입을 먼저 해주셔야 해요!
이메일 주소로 1분 정도면 가입하실 수 있습니다.
가입하시고 나면, 여러분의 개인 저장소 약 30 GB 가 제공되고 그때부터 모든 분석이 가능합니다.
잠깐 galaxy 서버의 외관을 설명드릴게요.
왼쪽은 여러분께서 사용하실 수 있는 여러 Tool 이 있습니다. (정말 많습니다)
tool 이 너무 많아서, 종류에 따라 여러 카테고리로 나눠져 있군요.
데이터 업로드, 텍스트 조작, RNA-seq 분석 등… 나중에 하나씩 살펴보시면서 원하시는 분석 도구가 있는지 확인해보세요!
가운데는 각 Tool 을 선택했을 때 확인하실 수 있는 툴의 여러 parameter, 입력 데이터 등을 조작하는 곳입니다.
왼쪽의 tool 을 하나하나 누를 때마다 가운데 화면이 바뀔 거에요.
오른쪽은 History 칸이 있습니다.
여기에는 여러분께서 진행하시는 여러 분석 단계들 및 중간 결과물들이 기록되는 곳입니다.
히스토리를 여러개 만들어서 프로젝트마다 관리할 수도 있어요.
그럼 분석을 시작해볼까요?
가장 먼저 해야할 일은 히스토리를 만드는 것입니다.
(회원가입을 하셨을 테니) 오른쪽 히스토리의 ‘+’ 버튼을 눌러서 한번 만들어 봅시다.
히스토리의 제목은 프로젝트 이름으로 해놔야 나중에 기억하기 쉽겠죠?
 |
|---|
| 저는 그냥 그날 날짜를 해당 히스토리의 제목으로 했습니다. |
쉽죠? 이제 업로드하는 데이터, 분석 결과 등은 이 히스토리에 쌓이게 될 겁니다.
데이터 업로드하기
그 다음에는 원하는 데이터를 업로드하시면 됩니다.
그 전에! 그럼 항상 데이터를 컴퓨터에 받은 다음 업로드 해야할까요?
물론 꼭 그런것은 아닙니다. 지금 여러분의 외장하드에 데이터가 있다면 그렇게 하시면 되지만요.
오늘은 GEO 에 있는 public data 를 다운로드 없이 galaxy 에서 사용하는 방법을 보여드릴게요.
그럼 먼저 GEO 에서 데이터를 찾아야겠죠?
이전 블로그를 보시면, GEO 에서 데이터를 찾는 방법을 보실 수 있을 거에요.
이번에 찾아야 할 데이터는 특정 GEO series 에서 SRR 리스트를 받아오는 작업입니다.
(* 잠깐. SRR 이 뭔가요?)
SRP, SRS, SRR 은 GEO sequence read archive (SRA) 라고 하는 저장소에 데이터들을 구분하기 위해 붙여둔 이름입니다.
SRP 는 GSE 에 해당합니다. 즉 각 연구 (study) 이고, 이는 여러 샘플의 묶음을 의미합니다.
SRS 은 GSM 에 해당합니다. 즉 각 샘플 (sample) 의 정보입니다.
SRX 는 각 실험 (Experiment) 에 대한 정보입니다.
SRR 은 각 Run 을 의미합니다. 한 개의 샘플을 여러 replicate 으로 시퀀싱을 진행했다면 한 샘플당 run 이 여러개가 있겠죠?
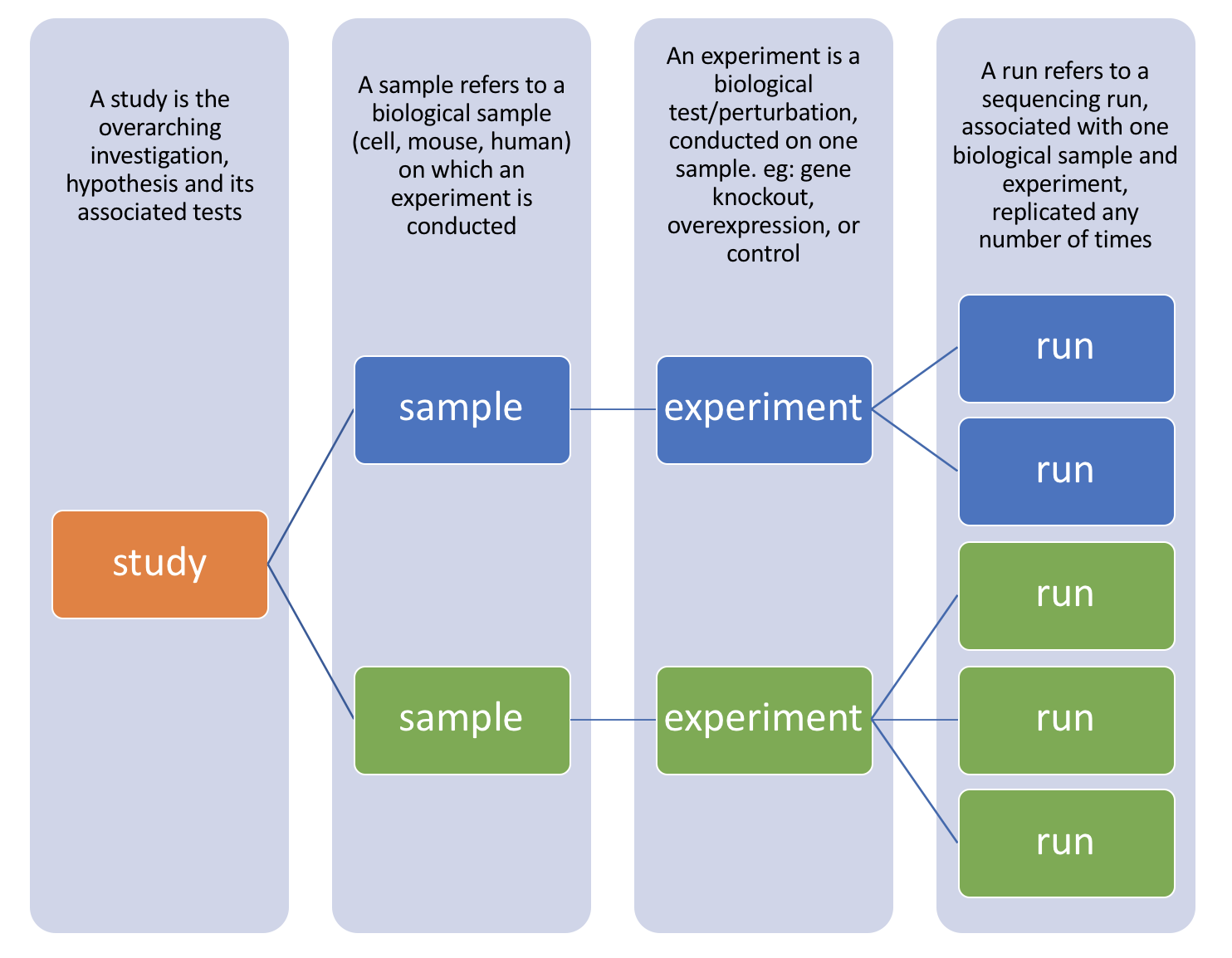 |
|---|
| 그림 출처: https://hbctraining.github.io/ |
우리에게 필요한 것은 한 연구에 포함된 sequencing run 목록, 그러니까 SRR 목록을 가져오는 것입니다.
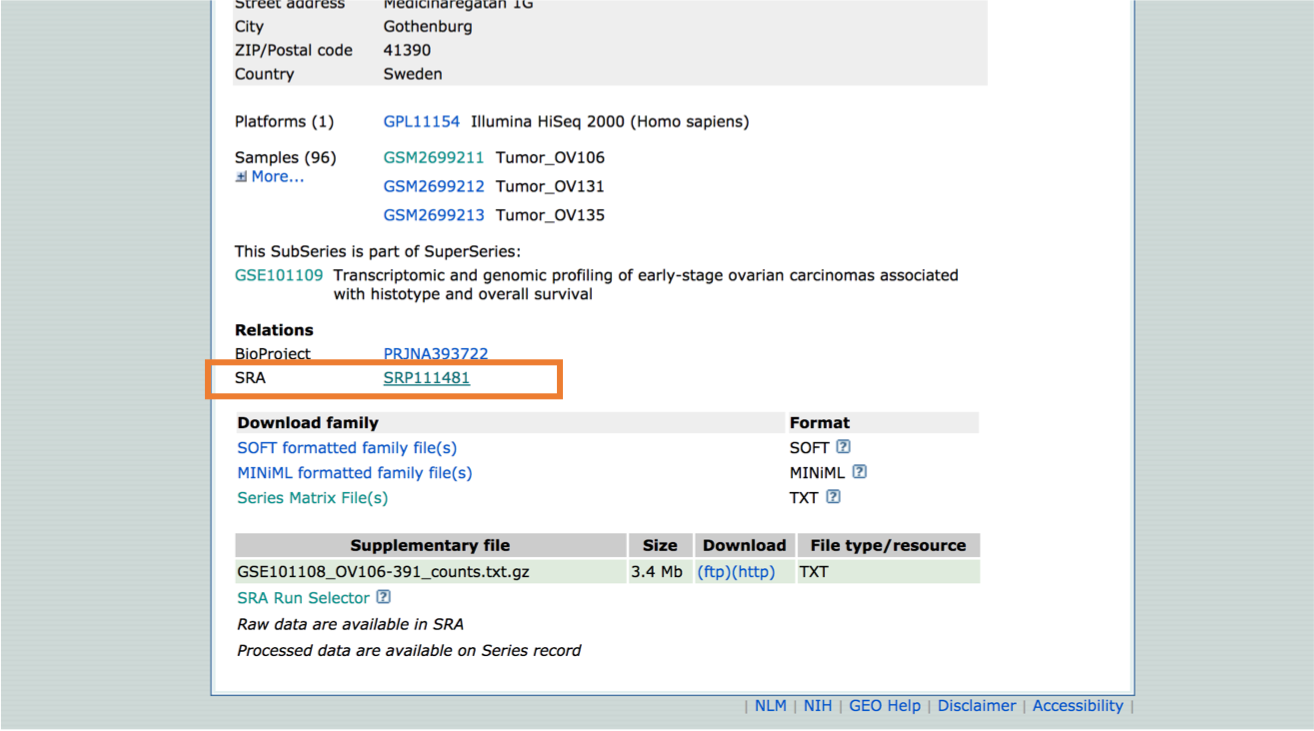
원하시는 GEO series 페이지에 들어가시면, 페이지의 아래쪽에서 SRP 정보를 찾으실 수 있습니다.
이걸 클릭해서 들어가 봅시다.
 |
|---|
| 이 시리즈는 이전 포스트에서 나왔던 Ovarian cancer study (GSE101108) 입니다 |
들어가보시면, SRA 사이트의 검색창이 나오고 아래에는 해당 SRP 에 관련된 샘플들이 결과로 나옵니다.
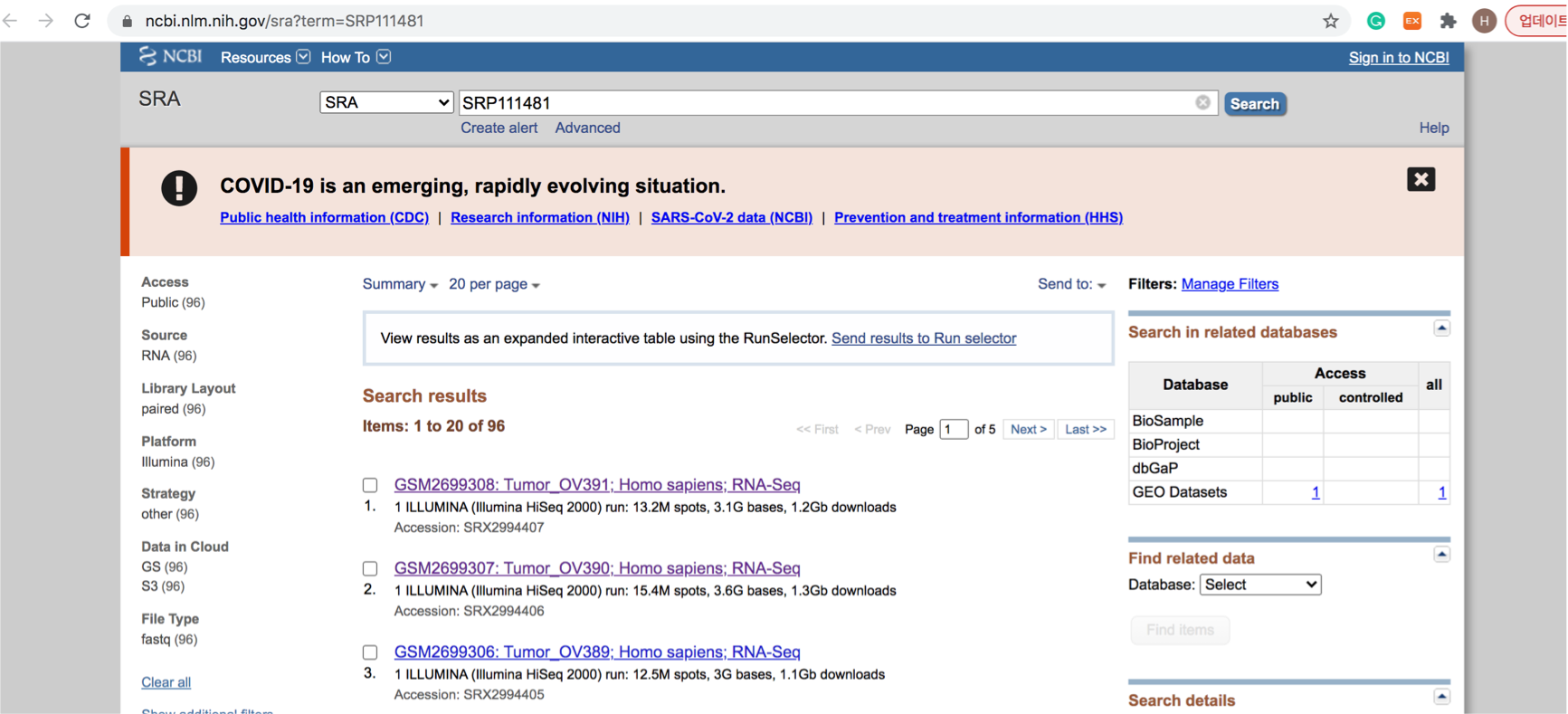 |
그런데 샘플이 96개나 되는군요. 한 페이지에 20개씩 밖에 안나오고 SRR 정보는 나오지도 않네요.
우리가 원하는건 모든 SRR 정보인데 말이죠.
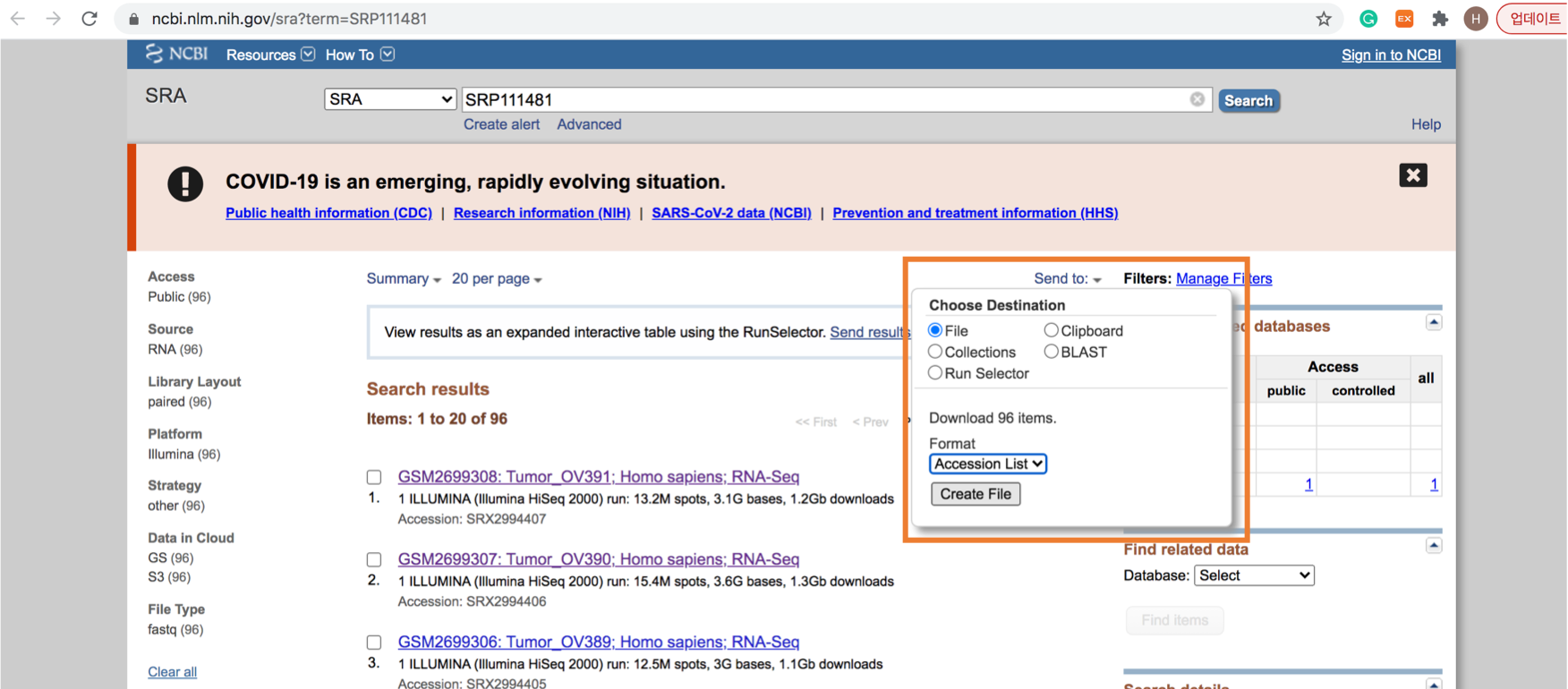
이를 한번에 받을 수 있는 방법이 있습니다.
아래 그림처럼 ‘Send to’ 를 누르시고, 여기서 ‘File’ 그리고 ‘Accession list’ 를 누르신 뒤 ‘Create File’ 을 누르시면 됩니다.
 |
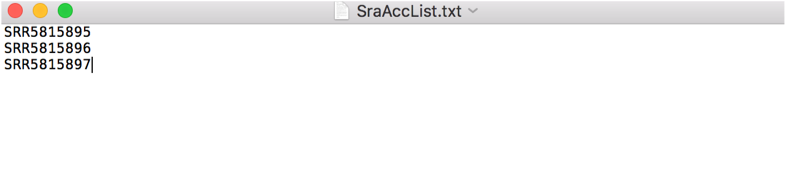
이렇게 하시면 파일이 하나 다운로드 될 텐데요.
열어보시면 우리가 원하던 것처럼 SRR** 라는 목록이 있을 겁니다!
 |
|---|
| 저는 예시로 만들고 싶어서 샘플을 3개만 남겨놨어요. 원래 96개가 다 있답니다. ^^; |
이렇게 하면 첫 단계가 끝납니다.
이제 정말로 Galaxy 에 데이터 업로드하기
SRR 목록을 만들었으니 galaxy 에 업로드 해볼까요?
왼쪽에 있는 Upload data 를 클릭해보세요.
그럼 창이 하나 뜨는데, 여기서 밑에 있는 Choose local files 를 누르고 아까 다운받은 SRR 목록 파일을 선택하세요.
그리고 오른쪽 아래의 ‘Start’ 를 누르시면 SRR 목록 업로드가 시작될 겁니다.
오른쪽 히스토리에 여러분의 첫 분석 단계가 시작된걸 보실 수 있습니다!
아래 그림에 과정을 나타내 보았습니다:
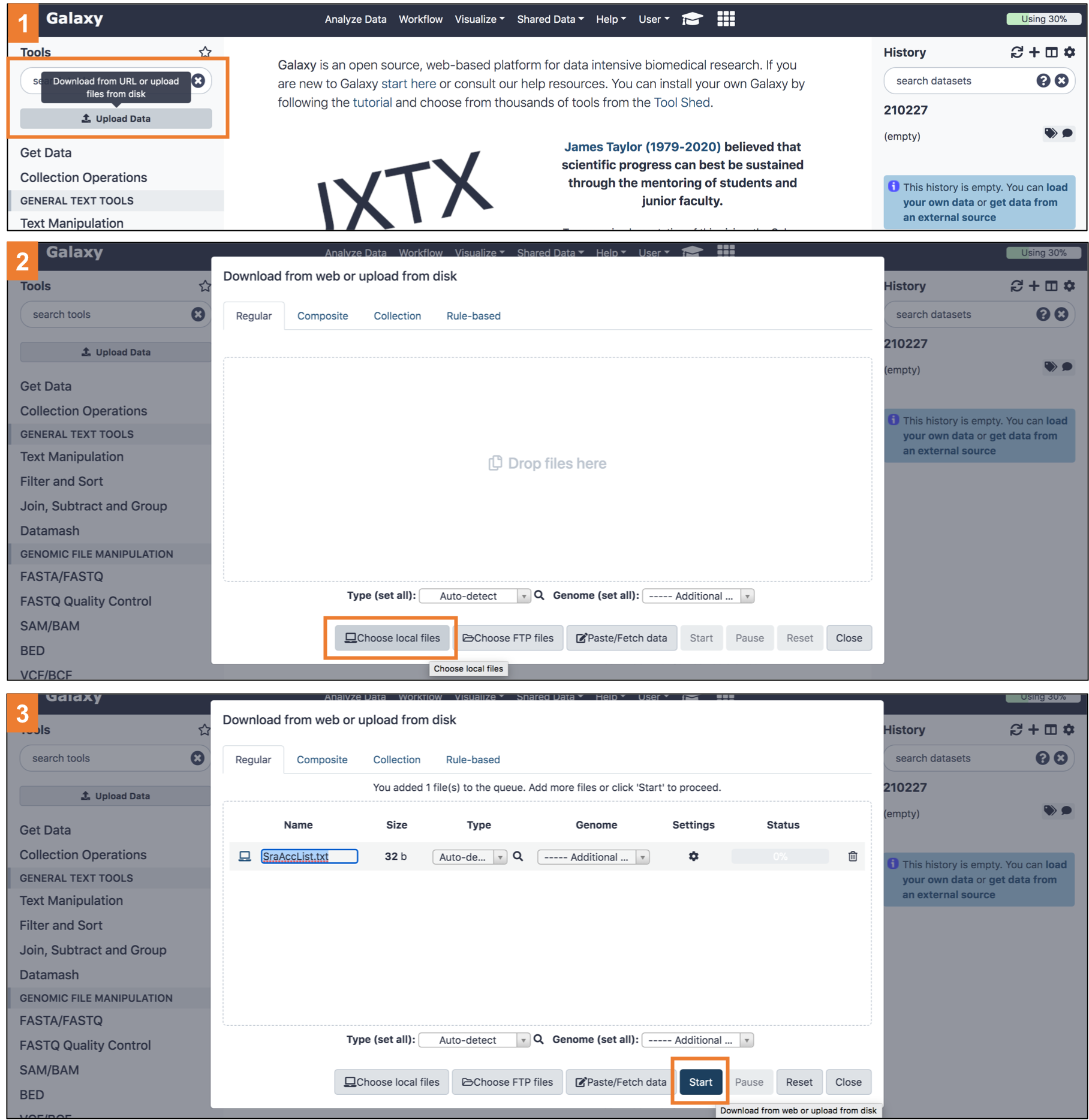 |
이렇게 하면 오른쪽 히스토리에 회색 (작업 전달) -> 주황색 (작업중) -> 초록색 (작업 완료) 순서로 나타날 거에요.
그리고 각 작업은 ‘번호’ 가 부여되니까, 나중에 이 번호를 참고해서 분석에 사용하시면 됩니다.
자, 그럼 업로드된 ‘목록’ 을 사용해 ‘실제 파일’ 을 받아볼까요?
왼쪽의 목록 중에 ‘Get Data’ 카테고리에서 ‘Faster Download and Extract Reads in FASTA/Q format from NCBI SRA’ 를 선택해보세요.
그럼 가운데에 여러 항목이 나오는데요, 여기서 다음 순서대로 진행하시면 됩니다.
- select input type : List of SRA accession, one per line 선택
- sra accession list : 방금 업로드한 SRA 목록 (히스토리의 번호 참조) 선택
- Execute 클릭!
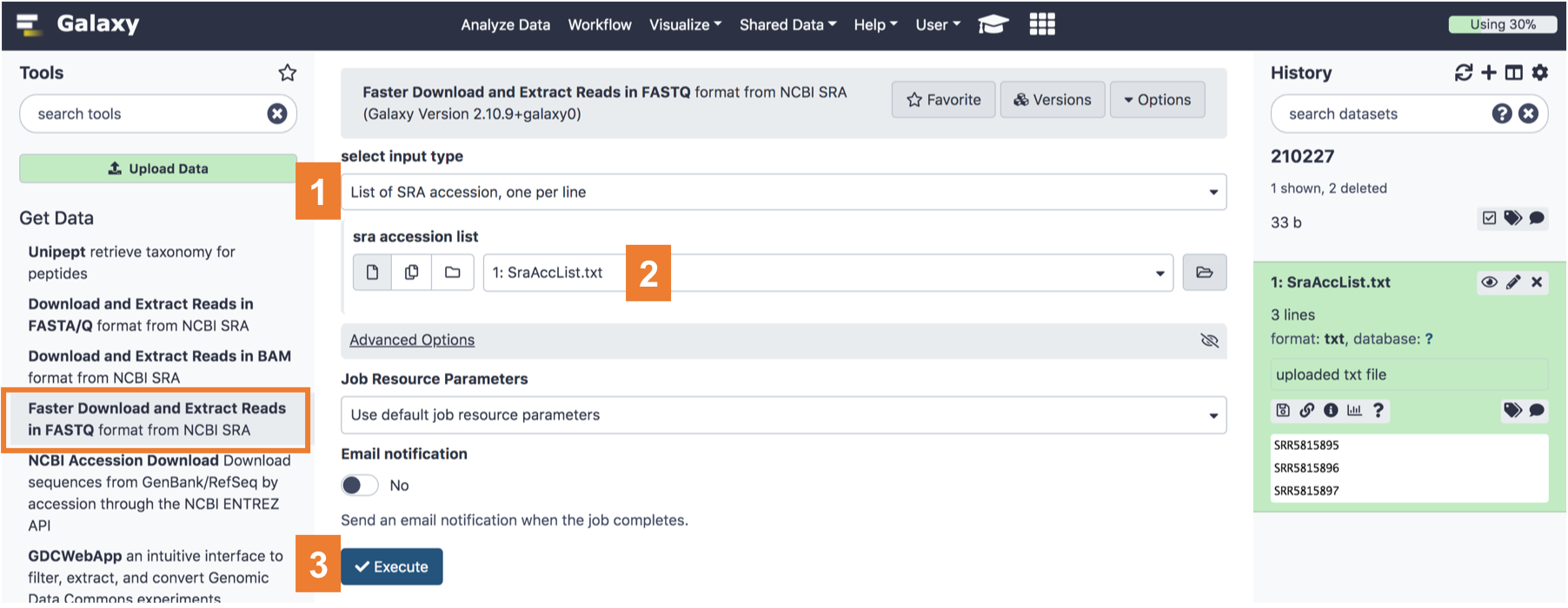 |
이렇게 하시면 galaxy 서버가 해당 SRR 들의 데이터를 받고, 알아서 fastq 라는 read data 로 변환해 준답니다.
이 과정은, 미국 GEO 서버에서 한국 컴퓨터로 데이터 받은 다음 다시 미국 galaxy 서버로 업로드할 필요가 없으니 좀 더 빠른 방법이라고 할 수 있습니다.
또한, .sra 파일 형식을 분석에 필요한 .fastq 형식으로 알아서 변환해주니 정말 편하죠.
파일이 업로드되는 시간은 꽤 오래 걸립니다.
엄청나게 많은 파일을 업로드하시는 거라면… 하루 이틀 기다리셔야 할지도 몰라요.
어쩔 수 없습니다. 시퀀싱 데이터들은 너무 크기가 커서 오래 걸릴 수밖에 없거든요.
저는 파일 3개 업로드 하는 데에 반나절 정도 걸렸습니다.
그러니 마음을 편히 가지시고, 하루에 한번 정도씩 galaxy 사이트에 들어가서 확인해보세요.
업로드가 다 됐다면, 다음과 같이 3 개의 파일이 생겼을 겁니다!
여러분이 업로드한 SRR 목록 중 single-end data 모음, paired-end data 모음, other data, 그리고 로그 파일입니다.
GSE101108 의 데이터들은 모두 paired-end data 이므로 아마 모든 데이터는 paired-end data collection 에 받아져 있을 거에요.
클릭해보시면, 업로드하신 파일의 목록이 뜰거구요, 각 SRR 을 클릭해보시면 forward, reverse data (paired-end data 이니까) 가 있을 거에요.
그리고 이를 각각 클릭해보시면 해당 fastq 파일의 스냅샷을 보실 수 있어요.
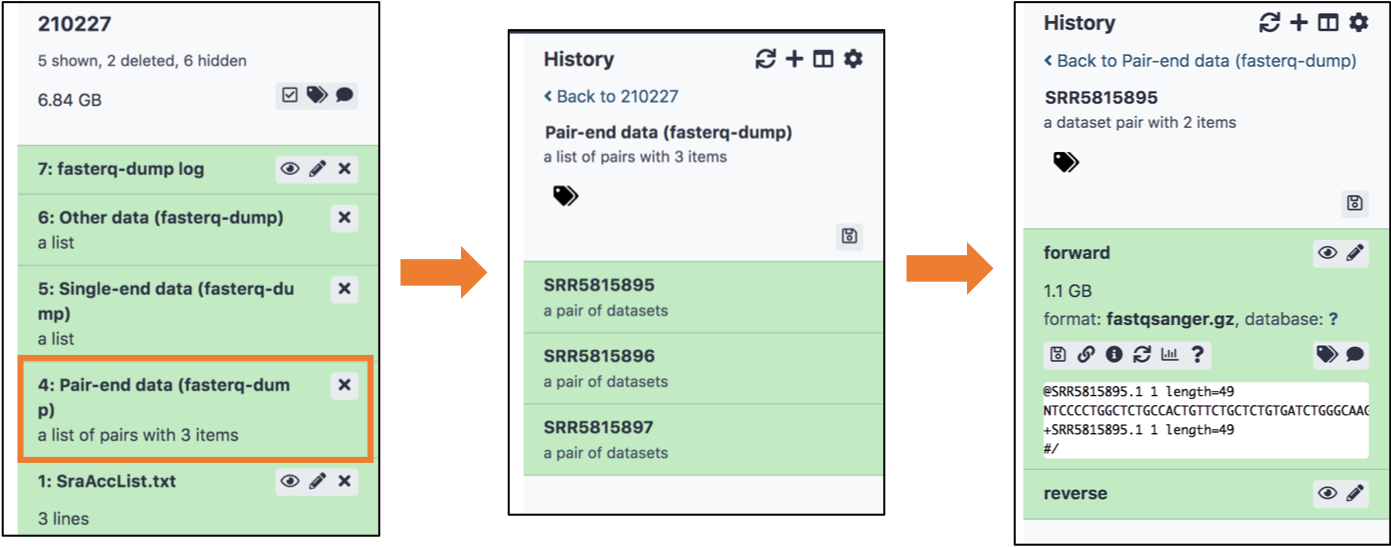 |
그럼 분석을 시작해볼까요?
RNA-seq 데이터를 분석하는 방법은 여러가지가 있습니다.
(* 여기서 ‘분석’ 이라고 말씀드렸는데, 사실은 raw data 에서 expression matrix 까지 만드는 과정을 말해요. ㅎㅎ)
기본적으로는, fastq 데이터를 reference genome 을 사용해 alignment 진행하고 이후 quantification 을 진행하는 거에요.
파일 형식으로 보면 fastq -> bam -> txt, tsv, 등등… 이 되는거죠.
여기서는 좀 더 빠르게 분석할 수 있는 방법을 사용해볼 거에요!
fastq 데이터를 reference genome 대신, reference transcriptome 을 사용해 mapping + quantification 을 한번에 하는 방법이죠.
커다란 genome 파일을 사용하지 않기 때문에 시간도 매우 적게 걸려요. (노트북 사양으로도 가능한 작업이라고 합니다)
그리고 결과 파일의 크기도 bam 이 아니기 때문에 매우 작답니다. cloud 에서 사용하기에 적절하죠.
이런 과정을 가능하게 해주는 tool 로는 여러가지가 있는데, 우리는 그 중 ‘kallisto’ 라는 걸 써볼 겁니다.
먼저, 준비물을 가져와 볼게요.
위에서 말씀드린 ‘reference transcriptome’ 파일이 필요합니다.
우리가 위에서 사용하는 데이터는 human sample 의 데이터이므로, reference 도 역시 human (homo sapiens) 데이터를 가져와야겠죠?
만약 다른 종의 데이터라면 아래 설명에서 종 정보만 다르게 하시면 나머지는 똑같이 분석하시면 됩니다!
reference transcriptome 는 RefSeq 이라는, NCBI database 를 사용해 받을거에요.
다음 링크로 가보시면 ftp 서버를 통해 여러 종의 다양한 시퀀스 정보를 제공하고 있습니다.
https://ftp.ncbi.nlm.nih.gov/refseq/
그리고 이 폴더구조에서 human reference transcriptome 정보는 다음 링크로 받으실 수 있어요.
https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh37_latest/refseq_identifiers/GRCh37_latest_rna.fna.gz
이 파일을 직접 받아서 아까처럼 업로드 해도 되는데요, 이번엔 ftp 링크를 통해 파일을 업로드하는 걸 보여드릴게요.
- 왼쪽의 Upload Data 를 누르고,
- 아래 메뉴 중 Paste/Fetch data 를 누른 뒤,
- 가운데 창에 위에 알려드린 reference transcriptome 파일의 링크를 복사해서 붙여넣으시면 됩니다.
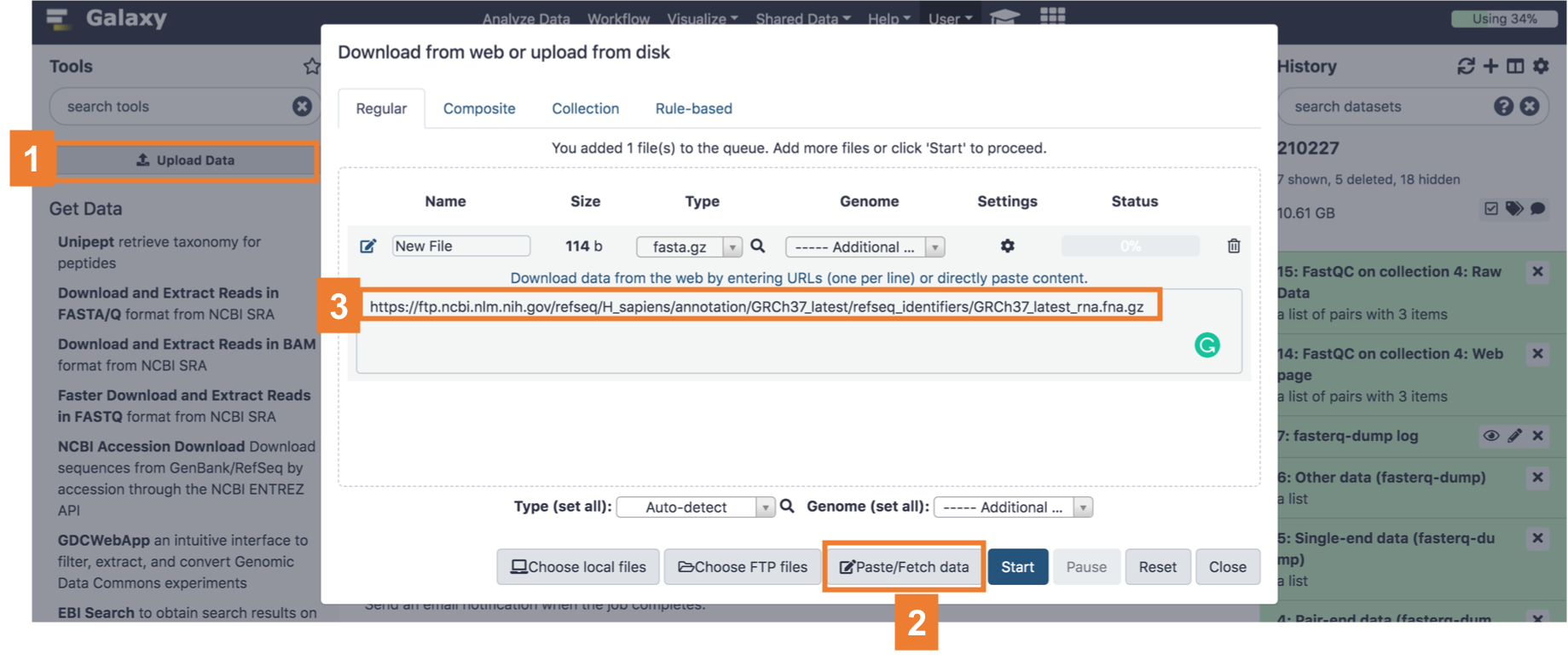 |
|---|
| 이 파일은 크기가 크지 않아서 몇분 내로 받아질 거에요. |
자, 업로드가 다 끝났다면 준비물도 있으니 분석을 해봅시다.
왼쪽의 검색창에 ‘kallisto’ 를 검색해보세요.
그럼 아래에 관련된 tool 목록이 나올 겁니다.
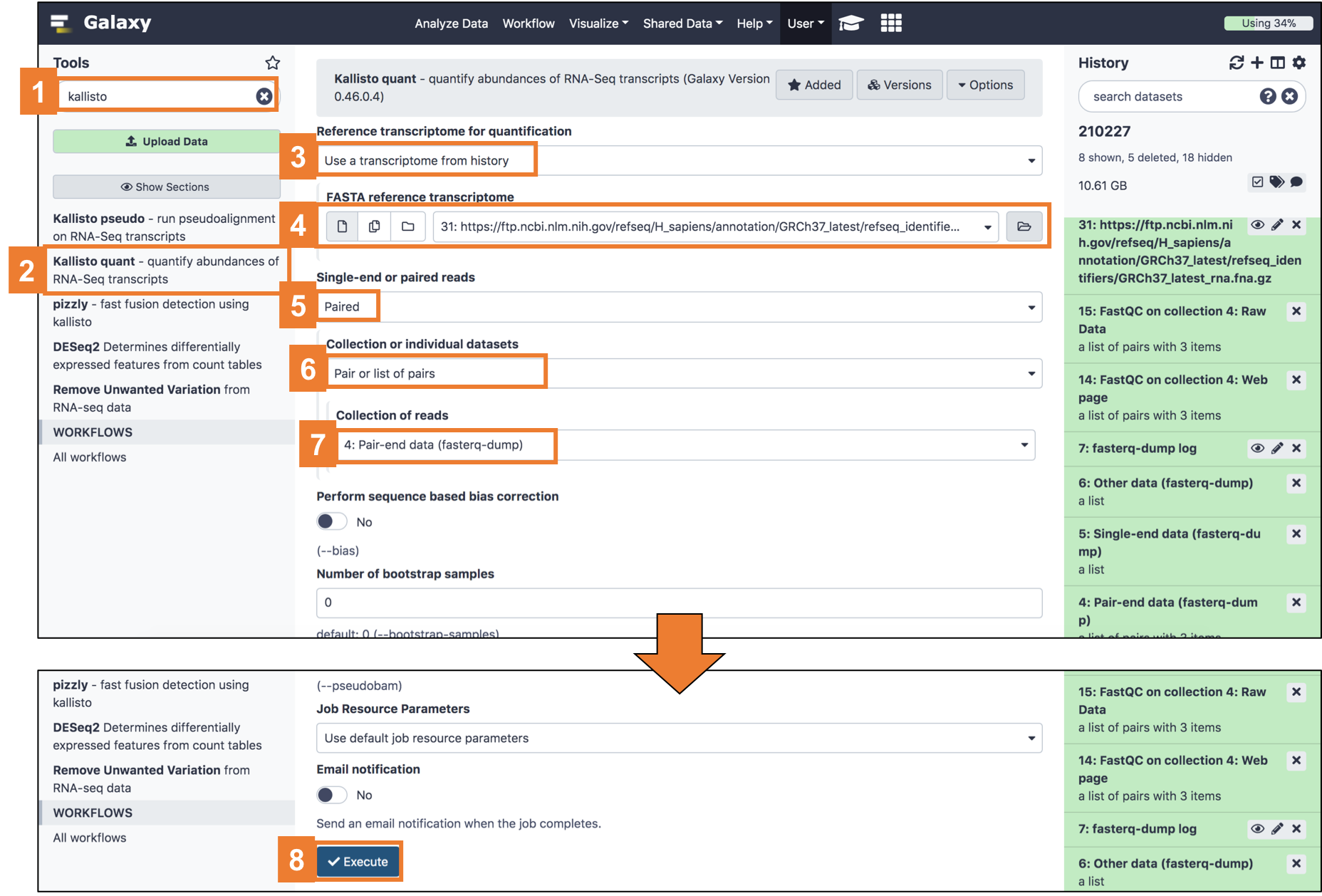
우리는 이중에 ‘Kallisto quant’ 라는 툴을 사용할 겁니다.
원래 kallisto 진행하려면 reference transcript indexing 을 먼저 해야하는데, 여기선 필요 없습니다.
사용을 쉽게 하기 위해서 이 작업까지 알아서 진행해주거든요.
아래와 같이 선택하시면 분석이 진행됩니다.
- kallisto 검색
- Kallisto quant 선택
- 우리가 reference transcriptome 을 업로드 했으니, 이를 사용하도록 Use a transcriptome from history 선택
- 아까 업로드한 transcriptome 파일을 선택 (히스토리 넘버를 잘 기억하세요!)
- RNA-seq 데이터가 paired-end 데이터이므로 paired-end 선택
- 여러 SRR 을 collection, 즉 묶음으로 업로드 했으니 Pair of list of pairs 를 선택
- 위에서 SRR 목록으로 가져온 RNA-seq 데이터 선택 (히스토리 넘버를 잘 기억하세요!)
- 이제 Excute 를 누르면 분석이 시작됩니다!
 |
이 분석은 생각보다 빠르게 될 겁니다. (빠르면 십분 이내로?)
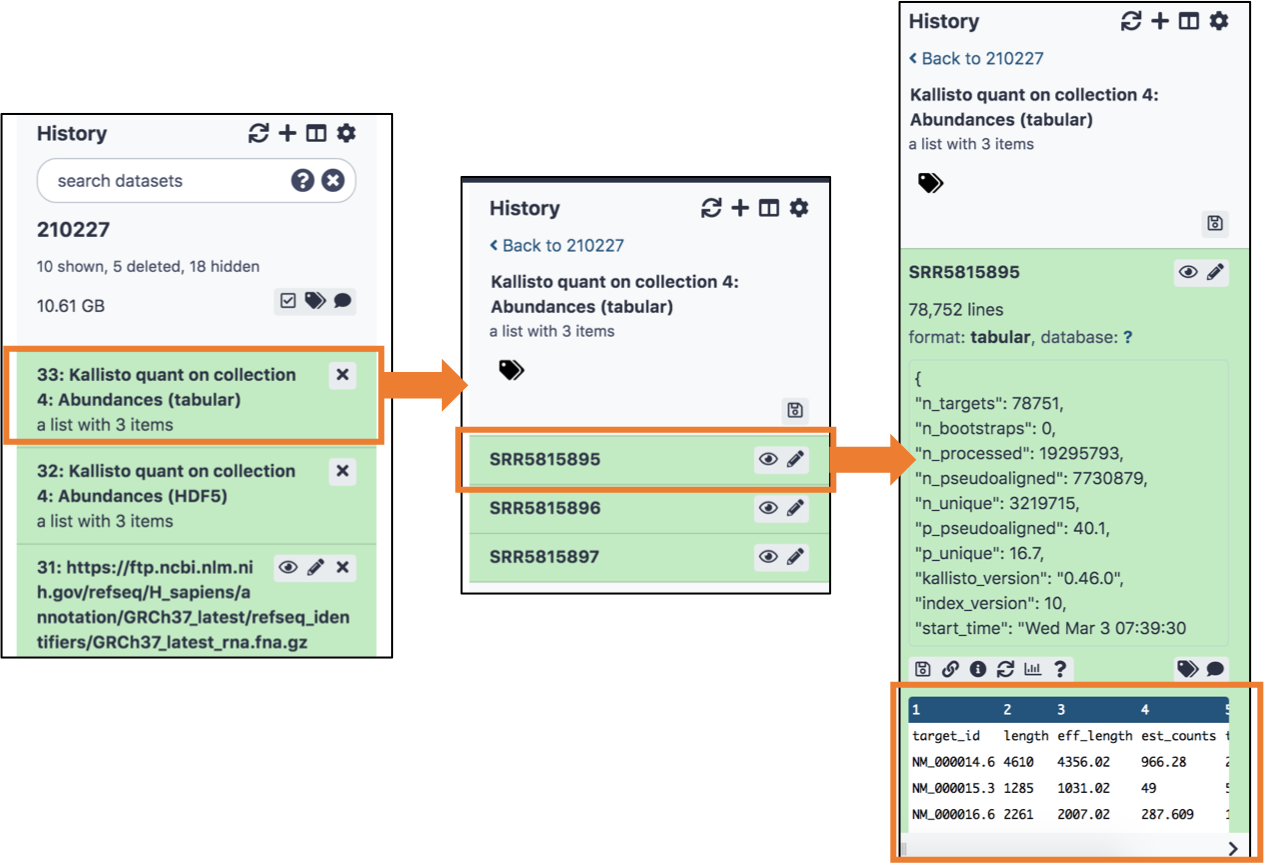
결과는 두 가지가 나오는데요, 아래 tabular 형식이 우리가 흔히 아는 표 형태의 파일입니다.
각 SRR 마다 파일이 하나씩 나오고, 마지막 column 에 ‘TPM’ 값이 나온답니다.
우리가 자주 보던 expression level 을 나타내는 값 중 하나죠?
이제 이 정보를 활용하기만 하면, RNA-seq 분석을 하실 수 있는거에요!
 |
|---|
| 결과 만들기 끝! |
그런데, 제가 생각한 형태가 아닌데요.
맞습니다. 사실 이 결과는 분석에 사용하기 쉬운 형태는 아니죠.
지금 여러분께서 기대하는 형태는 expression matrix 와 다른 점이 몇 가지 있습니다.
- expression level 이외의 데이터가 들어 있다는 것 (transcript length 등)
- Gene name 이 아니라 NM.001234 처럼 알 수 없는 이름으로 되어있다는 것 (이건 transcript ID 에요.)
- 결과 파일이 하나가 아니라 각 샘플당 파일 하나씩 여러개 파일로 나누어져 있다는 것
사실 이런걸 해결하는 작업이 생명정보학 전공자의 업무의 대부분을 차지한답니다.
보통 이런 작업은 코딩을 해서 진행하는데, 맨 처음에 그런거 없이 분석할 수 있는 방법을 소개해드린다고 했죠?
다행히도, Galaxy 사이트에서도 이 작업을 진행하실 수 있습니다!
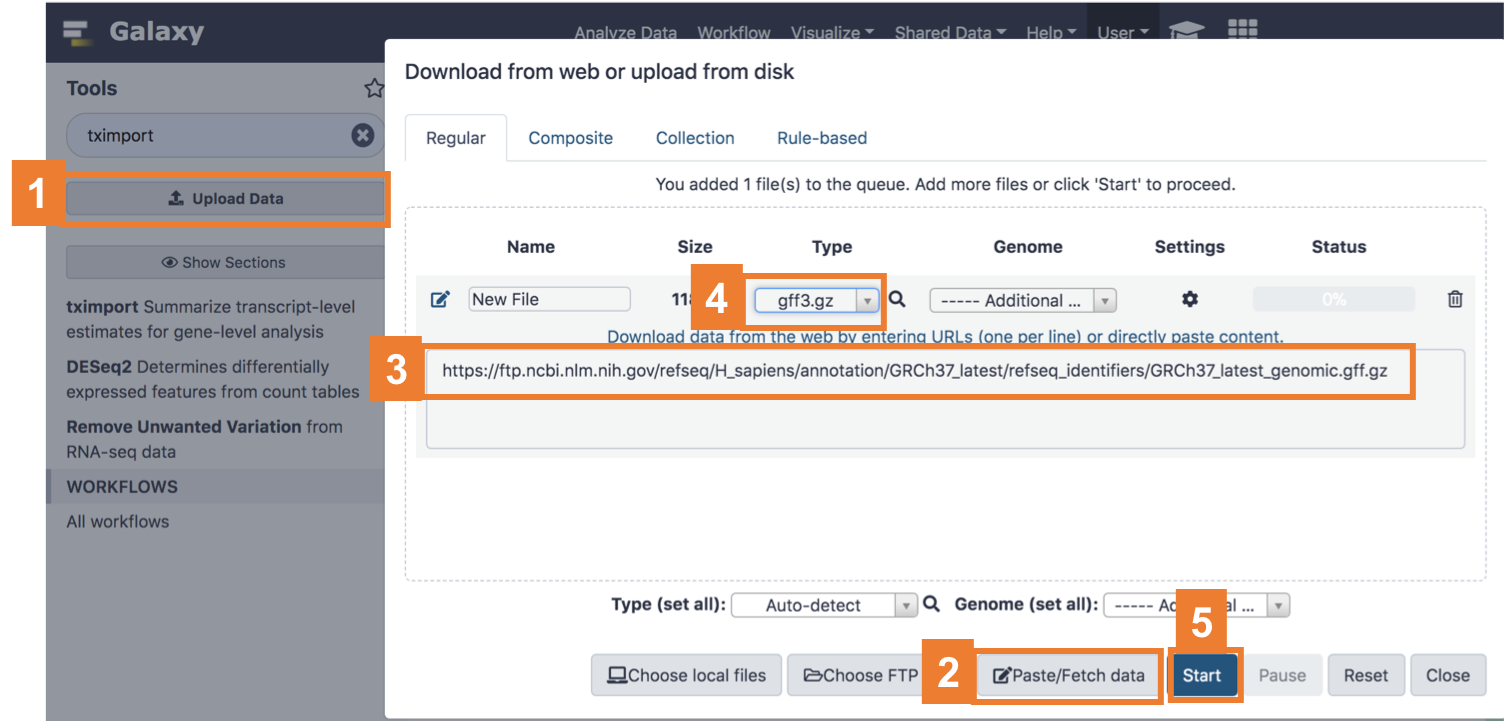
먼저 준비물이 필요한데요, gff 라는 파일을 받아와야 해요.
gff 형식이 무엇인지는 제 블로그의 다른 포스트를 보시면 된답니다 ㅎㅎ 설명 링크
이 gff 파일은 이번 작업에서 transcript 와 gene symbol 간의 관계를 만들 때 사용될 거에요.
아래 URL 을 사용하셔서, 아까처럼 link 를 통해 데이터 업로드하시면 gff 가 진행됩니다.
Refseq GRCh37 GFF link: https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh37_latest/refseq_identifiers/GRCh37_latest_genomic.gff.gz
- Upload data 선택
- Paste/Fetch data 선택
- URL 복사
- ‘Type’ 에서 gff3.gz 을 검색해서 선택
- ‘start’ 를 누르면 업로드가 시작됩니다.
 |
이렇게 하고나면 gff 파일이 업로드 된답니다.
준비물이 있으니 다음 단계를 진행해볼까요?
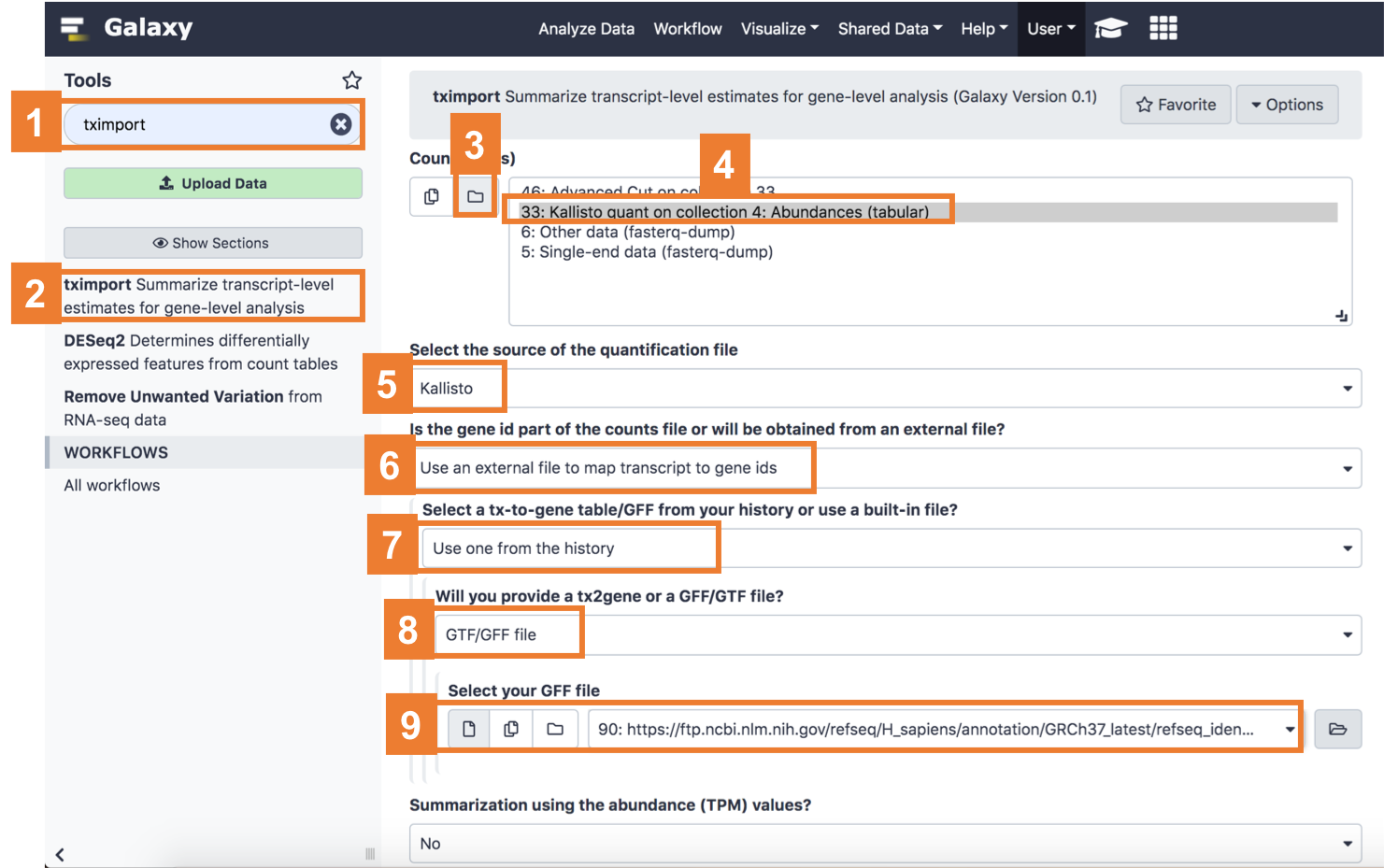
우리가 이번에 사용할 tool 은 tximport 라는 거에요.
이것은 transcript 별로 되어있는 RNA-seq 데이터를 Gene 단위로 묶어주는 작업을 해줍니다.
아래 순서대로 진행하시면 됩니다!
- 왼쪽 tool 에서 tximport 검색
- 검색 결과 중 tximport 선택
- ‘Dataset collections’ 선택
- 아까 만든 Kallisto quant (tabular) 결과물 선택 (히스토리 넘버를 잘 기억하세요!)
- quantification source 로 ‘Kallisto’ 선택
- ‘Use an external file to map transcript to gene ids’ 선택
- ‘Use one from history’ 를 선택
- ‘GTF/GFF’ 파일 선택
- select your GFF file 에서 아까 업로드한 GFF 파일 선택 (히스토리 넘버를 잘 기억하세요!)
- 이후 아래쪽에 있는 ‘Execute’ 을 누르면 진행됩니다.
 |
자, 이 작업을 완료하고 나면 ‘Gene level summarization’ 이라는 결과파일이 하나 생길겁니다.
이 파일이 여러분들께서 좀 더 작업하기 용이한 expression matrix입니다.
각 gene 의 expression level 이 sample 마다 TPM 으로 계산된 데이터입니다.
이 데이터를 활용해서 heatmap 도 그려볼 수 있고, differential expression 도 게산해보실 수 있어요.
히스토리의 결과에서 디스크 모양을 클릭하시면 결과를 다운받으실 수 있답니다.
결과는 .txt 파일이지만 이를 엑셀로 열어보시면 됩니다.
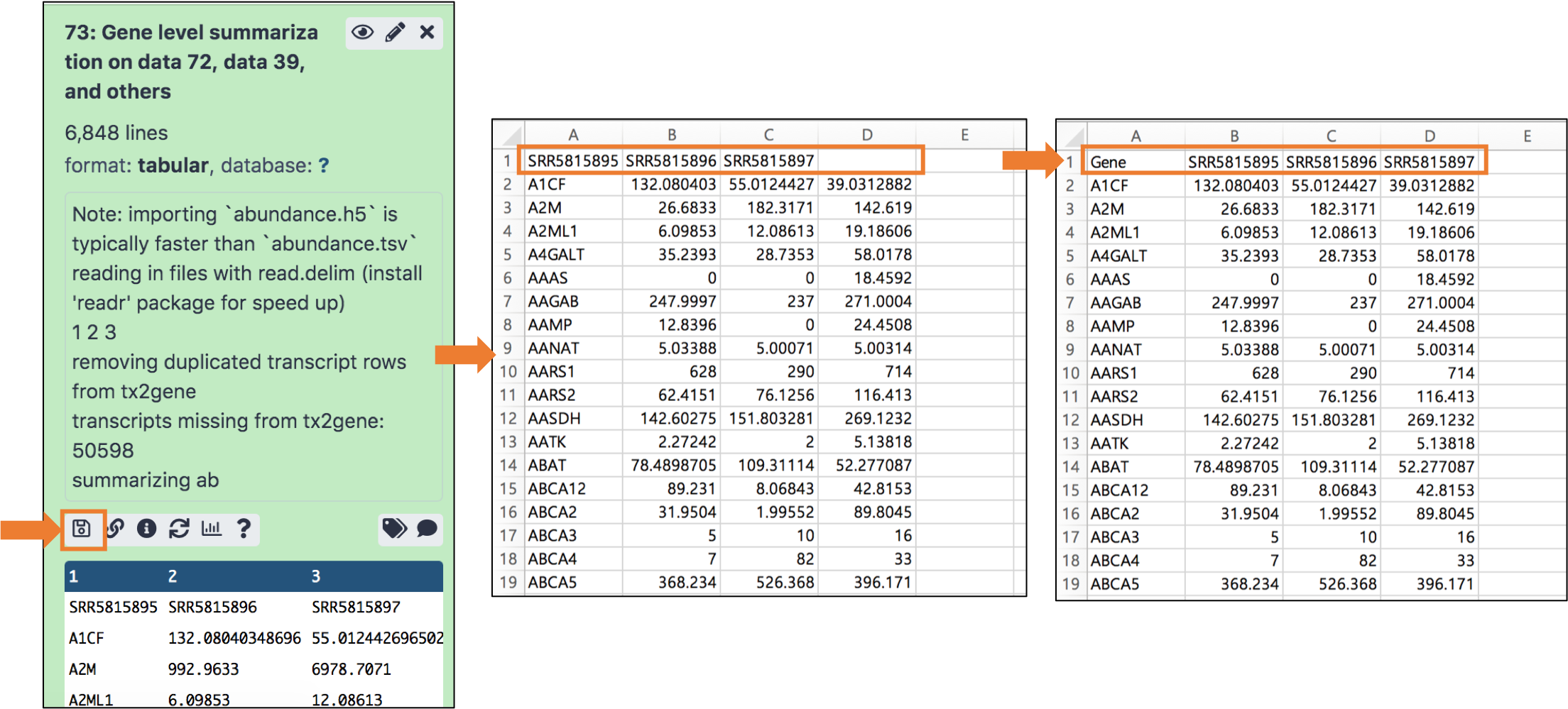 |
한 가지 주의해야 할 점이 있는데요, 이 파일을 받아서 엑셀로 열어보시면 맨 위쪽 샘플 이름칸이 하나씩 밀려있는 것을 볼 수 있을 거에요.
맨 오른쪽 열의 이름이 비어있죠?
이는 분석 결과가 R 을 사용해 만들어졌기 때문에 일어나는 어쩔 수 없는 일이에요.
받아보신 뒤 샘플 이름들을 하나씩 오른쪽으로 옮겨주시고, 첫 열의 이름을 Gene 으로 바꿔주세요.
네, 오늘은 RNA-seq 데이터를 raw data 에서 expression matrix 까지, 서버 없이 만드는 방법을 말씀드렸어요.
많은 분들께 유용했으면 좋겠네요.
사실, 이렇게 RNA-seq 분석을 하는 방법은 매우 다양하고, 그 상황에 맞게 만들어야 정확한 분석이 된답니다.
다음 시간에는 RNA-seq 결과를 통해서 Differentially Expressed Genes, 즉 DEG 을 찾아내는 방법과 찾아낸 DEG 을 사용해 어떤 분석이 가능한지를 보여드릴게요.
그럼 다음 시간에 만나요!