CPM, RPKM, FPKM, TPM 알아보기
안녕하세요 한헌종입니다.
이번 글에서는 RNA-seq 데이터에서 볼 수 있는 Expression value 에 대해 알아보겠습니다.
RNA-seq 데이터에 써있는 CPM, FPKM 등의 값을 사용하시면서 정확히 어떤건지 모르셨다면 이번 글이 도움이 되실거에요.
RNA-seq 에서 발현량이란?
RNA-seq 은 샘플에서 mRNA 를 추출하고 이를 cDNA library 로 만든 다음, 시퀀싱 기계로 읽어내는 실험방법 입니다.
시퀀싱 기계가 읽어낸 각 cDNA library 조각의 서열을 read 라고 부릅니다.
이렇게 읽은 각 read 서열이 genome 의 어디에서 온건지 컴퓨터로 알아내는 과정이 바로 alignment (혹은 mapping) 입니다.
이 과정을 간단히 그려보면 아래와 같아요.
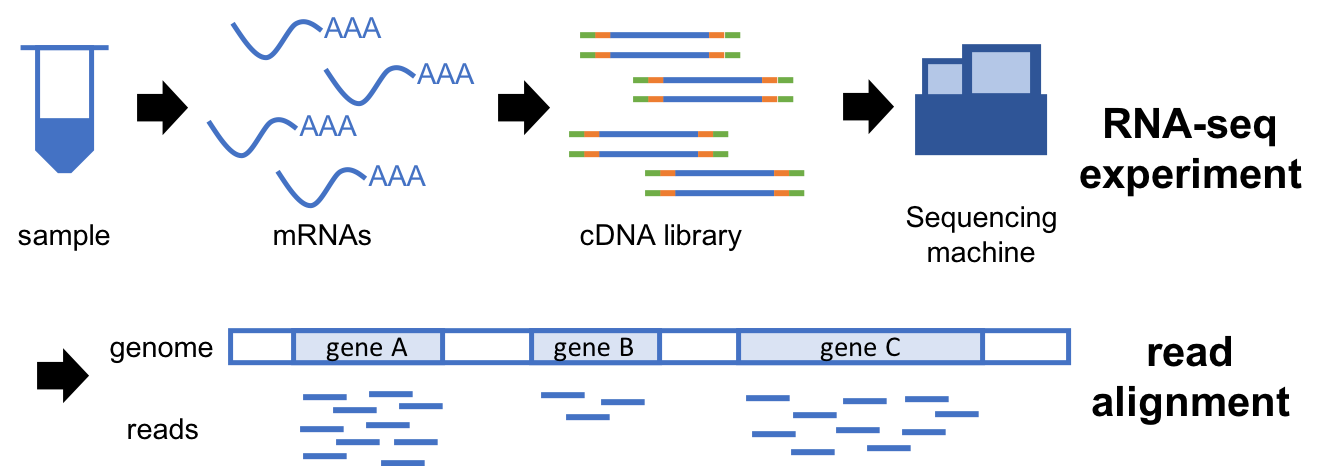 |
RNA-seq 에서 발현량이란 결국 각 유전자에서 발견된 read 의 개수를 의미합니다.
그림의 아래쪽을 보면 gene A, gene B, gene C 에 여러 개의 read 가 발견된 것을 보실 수 있죠?
여기서 중요한 질문이 나옵니다: 어떤 유전자의 발현량이 가장 많을까?
이는 계산법에 따라 다른 결과를 가져올 수 있습니다.
아래에서 CPM, RPKM, FPKM, TPM 을 알아본 뒤에 이 질문의 답을 알아보겠습니다.
CPM
CPM 은 counts per million mapped reads 의 약자입니다.
가장 간단한 발현량 계산법으로, 각 유전자 (혹은 transcript) 에 발견된 read 개수로 발현량을 평가하는 거죠.
이 때, 실험 결과로 나온 전체 read 개수가 애초에 많았다면 CPM 이 커지게 되므로, 이를 전체 read 개수로 나눠주는 ‘보정’을 해줍니다.
그런데 보통 전체 read 수가 매우 크니까 (보통 몇천만 개) read 개수를 이걸로 나눠버리면 너무 작은수가 될거에요.
그래서 million, 즉 백만이라는 숫자를 곱해서 “보기좋은 숫자”로 만들어주면 드디어 CPM 이 완성됩니다.
어떤 유전자의 read count 를 count, total read count 를 total 이라 하면 CPM 은 아래와 같습니다.
CPM = count * 1,000,000 / total
유전자가 A, B, C 세 개 있고, 각각 read count 가 5, 5, 10 인 경우
CPM 은 다음과 같이 나타낼 수 있습니다:
| Gene | read counts | CPM |
|---|---|---|
| A | 5 | (5 * 106) / 20 |
| B | 5 | (5 * 106) / 20 |
| C | 10 | (10 * 106) / 20 |
(이건 유전자 3개짜리 예시라서 106 을 곱해주는 보정이 의미가 없네요)
즉, 발현량은 C 가 제일 많고, A 와 B 가 같네요.
그런데 단순히 read count 로 세어버리면 오류가 생기지 않을까요?
그래서 나온 것이 RPKM, FPKM 입니다.
RPKM, FPKM
RPKM 먼저 설명드리겠습니다.
RPKM 은 reads per kilobase of transcript per million mapped reads 의 약자입니다.
CPM 과 다른점은, 바로 transcript 의 길이로 보정해준다는 점이죠.
예를 들어 길이가 1,000 basepair 인 transcript A 있고, 길이가 100,000 basepair 인 transcript B 가 있다고 해봅시다.
두 transcript 에서 똑같이 reads 가 50개씩 발견되었다면, 과연 두 transcript 의 발현량이 똑같은 걸까요?
왠지 아닌 것 같죠?
transcript A 보다 B 가 훨씬 더 길기 때문에, B 에서 더 많은 read 가 발견되어야 발현량이 동등하다고 할 수 있겠죠.
이런 시각을 적용한 계산법이 바로 RPKM 입니다.
발현량을 구할 때 각 transcript 에서 발견된 read 개수를 transcript 길이로 나눠서 보정해주는 작업이 추가된에요.
그리고 역시나 transcript 길이가 꽤 큰 값이니까 너무 작아지지 않게 1,000 을 곱해줘서 “보기좋은 숫자”로 만들어줍니다.
transcript 의 길이를 length 라고 했을 때, RPKM 은 다음과 같이 계산됩니다.
RPKM = (count * 1,000 * 1,000,000) / (length * total)
결과적으로 길이와 전체 개수로 나눈 뒤 109 을 곱해주는 거에요.
유전자가 A, B, C 세 개 있고, 각각의 길이가 10, 5, 20 인데 read count 가 각각 5, 5, 10 인 경우,
RPKM 은 아래처럼 표로 나타낼 수 있습니다:
| Gene | Length | read counts | RPKM |
|---|---|---|---|
| A | 10 | 5 | (5 * 109) / (10 * 20) |
| B | 5 | 5 | (5 * 109) / (5 * 20) |
| C | 20 | 10 | (10 * 109) / (20 * 20) |
이렇게 계산해보니, 발현량은 B 가 제일 많고, A 와 C 가 같군요.
그렇다면 FPKM 은 뭘까요?
FPKM 은 RPKM 과 같다고 보시면 됩니다.
차이점은 RPKM 은 single-end data 에서 쓰이고, FPKM 은 paired-end data 에서 쓰인다는 점이죠.
paired-end data 에서는 하나의 cDNA library 를 양쪽에서 읽어 두 개의 read 가 만들어지고, 이 두 개의 read 가 한 쌍을 이룹니다.
이렇게 쌍을 이루는 reads 를 합쳐서 fragment 라고 하죠.
즉 FPKM 은 read count 대신 fragment count 를 사용한 것 뿐입니다. 계산식도 같습니다.
약자도 거의 비슷합니다. FPKM = fragments per kilobase of transcript per million mapped reads
이렇게 계산한 결과를 보면 꽤나 정교하게 보정된 값인 것처럼 보입니다.
그런데 이렇게 구해진 값이 과연 가장 정확한 발현량 값일까요?
이런 의문을 해결하기 위해 만들어진 단위가 바로 TPM 입니다.
TPM
TPM 은 RPKM, FPKM 과 매우 유사하지만, 계산하는 순서가 조금 다릅니다.
그 목적이 “샘플 간 비교를 더 정확히 하기 위해서” 라는 것을 기억하세요.
(물론 그렇다고 RPKM, FPKM 으로 샘플간 비교를 하면 안된다는 뜻은 아닙니다)
TPM 은 먼저 read count 를 길이로 보정한 뒤, (total read count가 아닌) 이렇게 보정된 값의 총 합으로 각각을 나누는 방법입니다.
조금 복잡해지니 표로 한번 설명해볼게요.
먼저 3개의 유전자가 있다고 가정해봅시다: A, B, C
그리고 각각 길이가 10, 5, 20 이라고 가정합니다.
read count 가 각각 5, 5, 10 이라고 하면 RPKM 과 TPM 은 다음과 같습니다.
(비교를 위해서 109 로 보정해주는 건 잠깐 생략할게요.)
| Gene | Length | read counts | RPKM | read counts / length | TPM |
|---|---|---|---|---|---|
| A | 10 | 5 | 5 / (10 * 20) = 0.025 |
0.5 | 5 / (10 * 2) = 0.25 |
| B | 5 | 5 | 5 / (5 * 20) = 0.05 |
1 | 5 / (5 * 2) = 0.5 |
| C | 20 | 10 | 10 / (20 * 20) = 0.025 |
0.5 | 10 / (20 * 2) = 0.25 |
read count 를 length 로 나눈 값은 각각 0.5, 1, 0.5 이구요,
이를 모두 합하니 2가 되네요.
따라서 각각의 read count 를 길이 뿐 아니라 “2”로도 나누어주면 바로 TPM 이 되는 겁니다.
RPKM 과 TPM 이 어떻게 다른가요?
이렇게 해놓고 보니, RPKM 과 10배 차이 난다는 것 빼고는 별다른 차이가 없어 보이죠.
하지만 TPM 의 목적이 “샘플 간 비교를 더 정확히 하기 위해서” 라고 했죠?
그러니 이번엔 두 샘플의 값을 비교해보도록 합시다.
아까와 똑같이 유전자 A, B, C 가 있고, 각각 길이가 10, 5, 20 이라고 가정합시다.
그리고 sample 1 에서는 read count 가 각각 10, 5, 10 이고, sample 2 에서는 각각 10, 0, 30 이라고 가정해봅시다.
(여기서도 비교를 위해서 109 로 보정해주는 건 잠깐 생략할게요.)
| Gene | Length | sample 1 read count |
sample 2 read count |
sample 1 RPKM |
sample 2 RPKM |
sample 1 TPM |
sample 2 TPM |
|---|---|---|---|---|---|---|---|
| A | 10 | 10 | 10 | 0.04 | 0.025 | 0.4 | 0.4 |
| B | 5 | 5 | 0 | 0.04 | 0 | 0.4 | 0 |
| C | 20 | 10 | 30 | 0.02 | 0.038 | 0.2 | 0.6 |
Count 로 보면 gene A 는 sample 1 에서 10, sample 2 에서 10 으로 동일합니다.
gene C 의 경우 sample 1 에서 10, sample 2 에서 30 으로 세 배 증가했군요.
RPKM 으로 보면 gene A 는 sample 1 에서 0.04, sample 2 에서 0.025 가 되어서 줄어들었네요.
gene C 는 sample 1 에서 0.02, sample 2 에서 0.038 로 두배 좀 안되게 늘었습니다.
TPM 으로 보면 gene A 는 sample 1 에서 0.4, sample 2 에서 0.4 로 동일합니다.
gene C 는 sample 1 에서 0.2, sample 2 에서 0.6 으로 세 배 늘어난 것으로 나오는군요.
자, 과연 어떤게 정확한 비교일까요?
여기서 TPM 중요한 특징이 하나 나옵니다.
바로 각 샘플의 TPM 총 합이 모두 동일하다 는 점이에요.
즉, sample 1 의 TPM 합 = 0.4 + 0.4 + 0.2 = 1 이고, sample 2 의 TPM 합 = 0.4 + 0 + 0.6 = 1 로써 동일하다는 겁니다.
‘샘플 간 비교를 더 정확히 하기 위해서’ 라는 말은 바로 이를 나타나는 것입니다.
각 샘플의 유전자 발현량의 총 합을 동일하게 만든 값이기 때문에, 특정 샘플에서만 나타날 수 있는 편향을 제거했다고 볼 수 있죠.
요약하자면 “TPM 은 유전자 길이 및 값의 총 합으로 보정했을 뿐 아니라, 각 샘플의 총 합을 동일하게 해서 샘플 간 비교를 더 정확히 할 수 있는 값”이라고 할 수 있겠네요.
네, 오늘은 CPM, RPKM, FPKM, TPM 에 대해서 알아봤습니다.
이중 어떤 값을 사용하셔도 무방합니다만, 더 정확히 알고 쓰면 좋겠죠?
생각보다 쉬운 수식이기 때문에, 여러분이 만약 수식을 정확히 알고 있고 데이터만 있다면 엑셀로 이를 계산하는 것도 불가능한 일은 아닙니다. (데이터가 너무 커서 문제지만…)
RNA-seq 분석하실 때 도움이 되시길 바랄게요.
그럼 다음 시간에 만나요!